MS and MS/MS
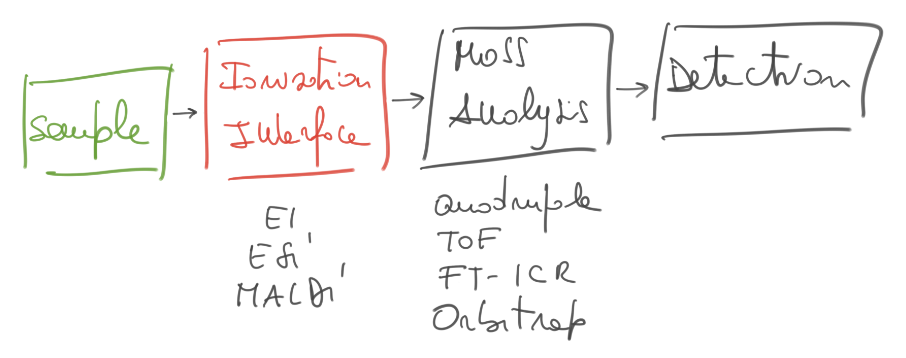
Structure of a Mass Spectrometer
Some basic concepts…
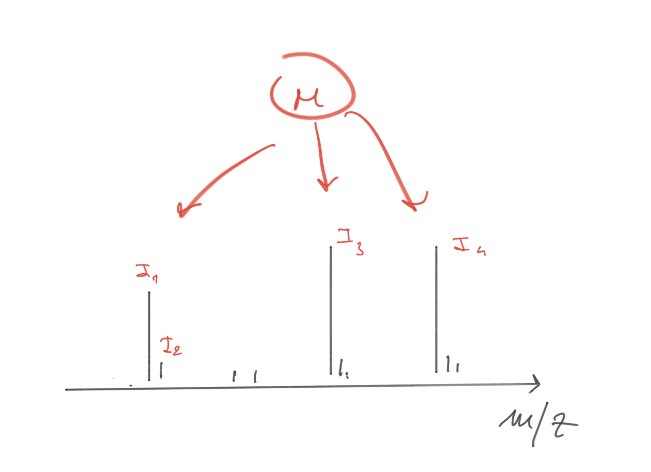
A simple mass spectrum is represented by a two dimensional bar plot:

Some basic concepts…
Usually each compound produce >1 ion
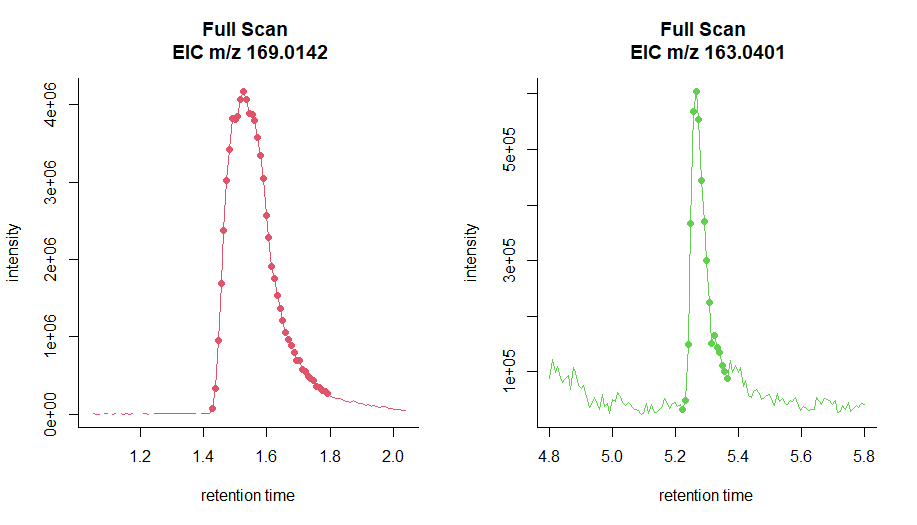
Points-per-peak
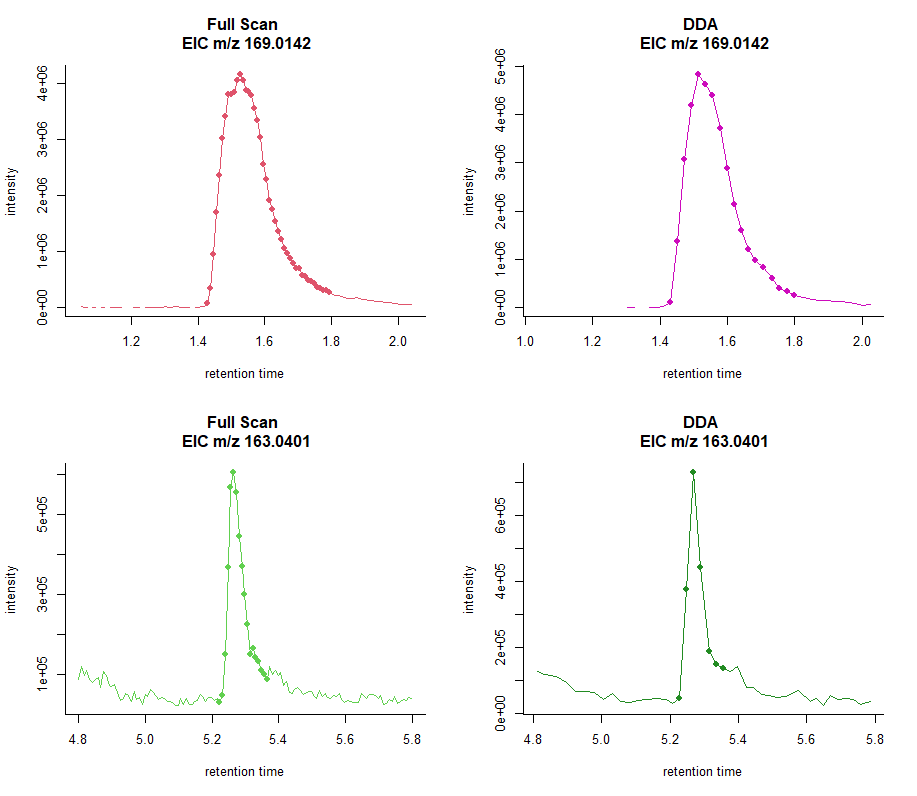
Enough data points per chromatographic peak
(i.e., at least 7–8)

Points-per-peak
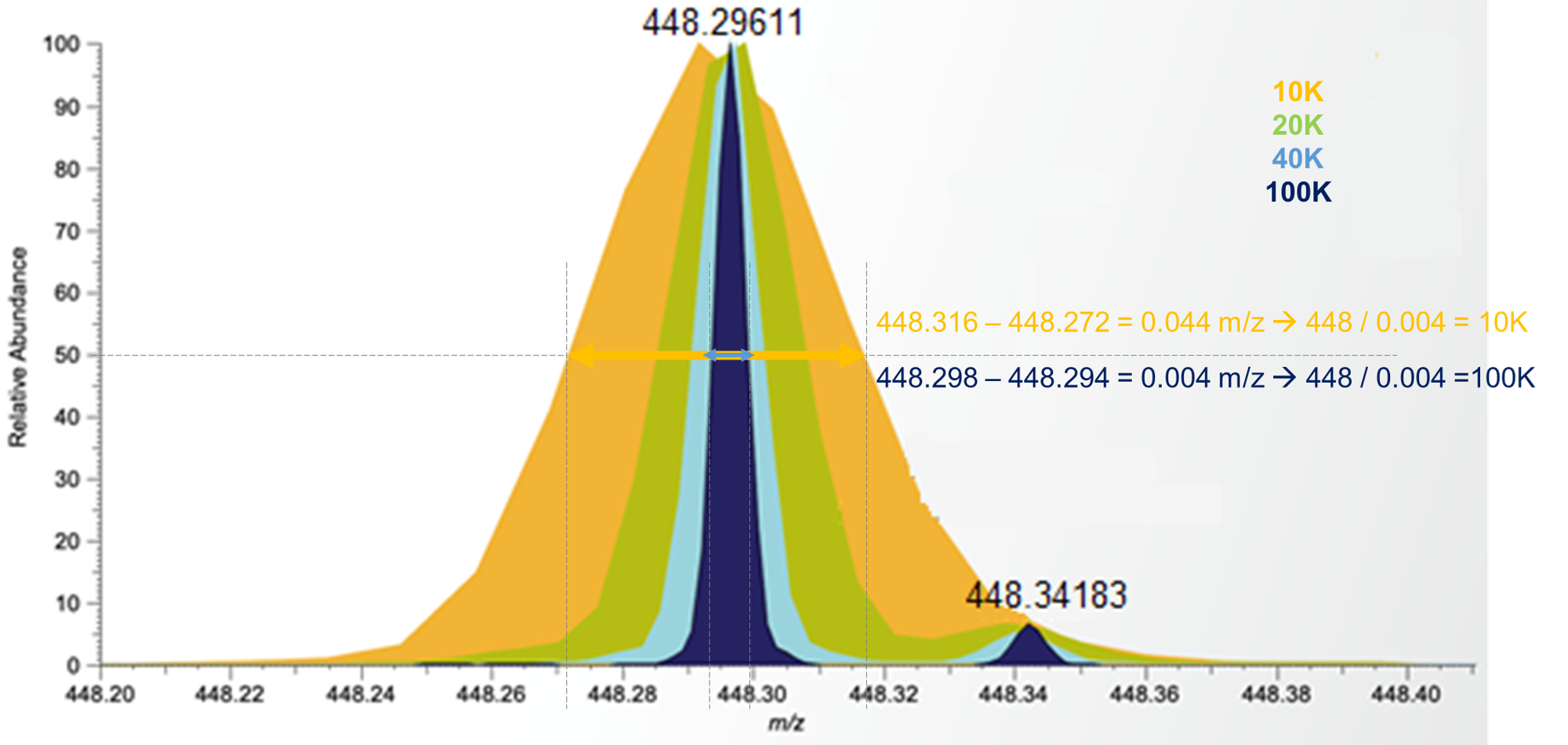
Resolving power
RP = M / d(M)
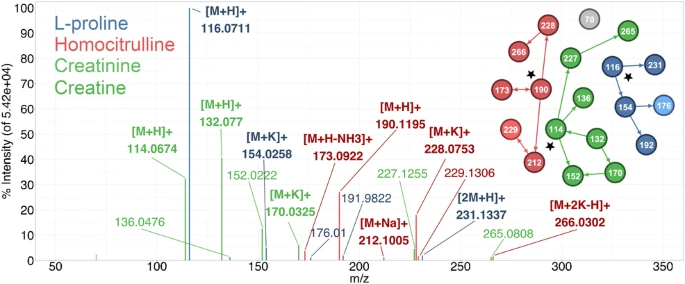
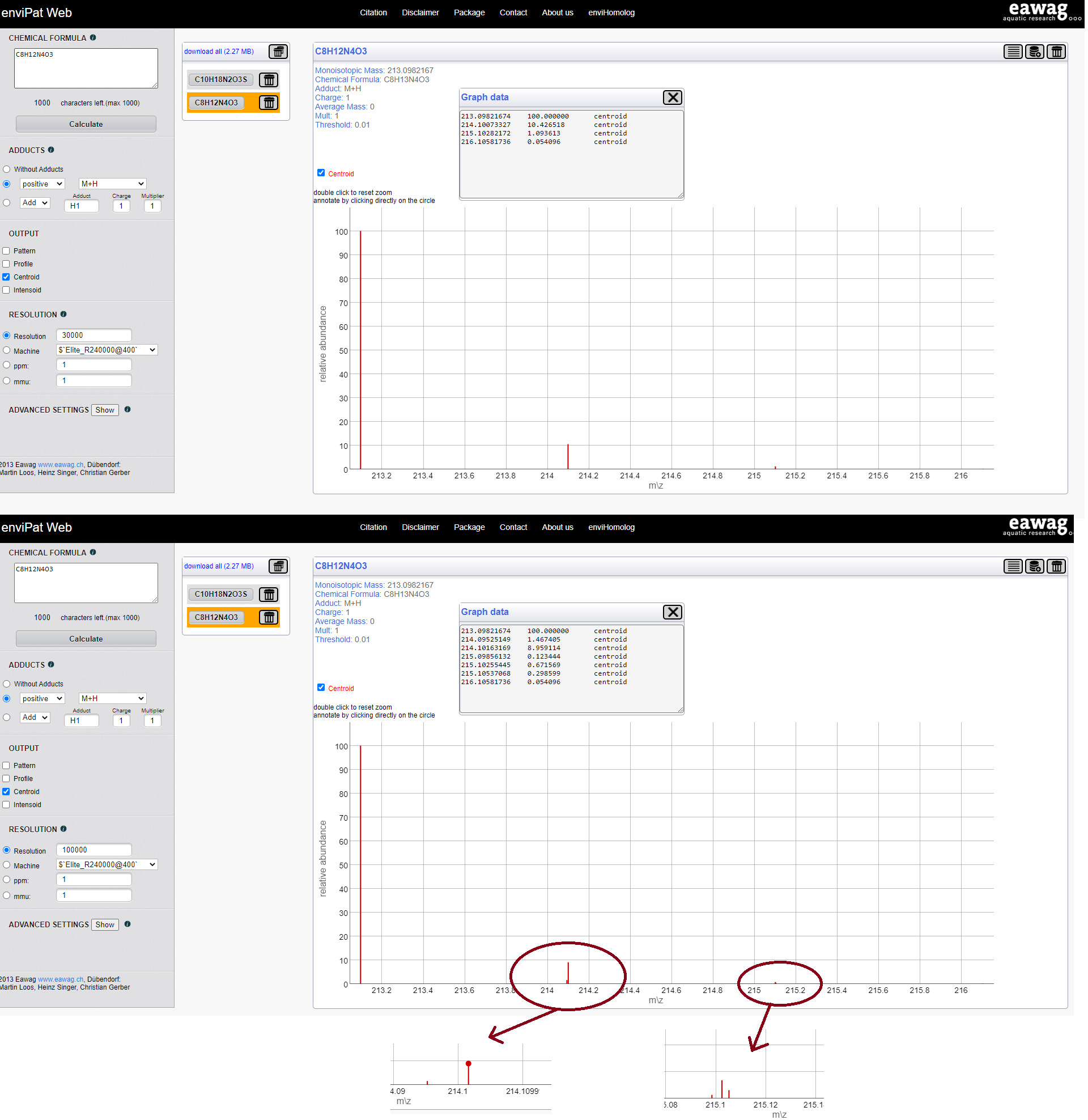
Isotopic patterns
Theoretical isotopic pattern calculator: enviPat
Check examples with: C7H6O5 and C8H12N4O3
Mass resolution & Isotopic patterns
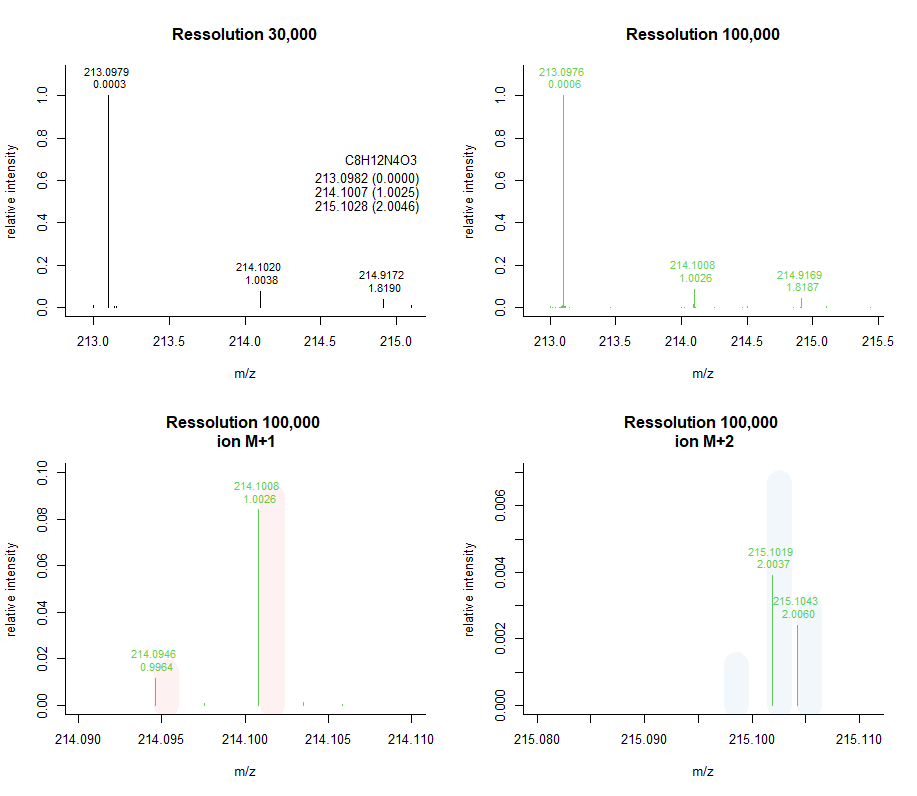
Mass resolution & Isotopic patterns
| Isotope | d(Mass) |
|---|---|
| C(12) | 0.000000 |
| C(13) | 1.003355 |
| N(14) | 0.000000 |
| N(15) | 0.997035 |
| O(16) | 0.000000 |
| O(17) | 1.004216 |
| O(18) | 2.004244 |
| H(1) | 0.000000 |
| H(2) | 1.006277 |
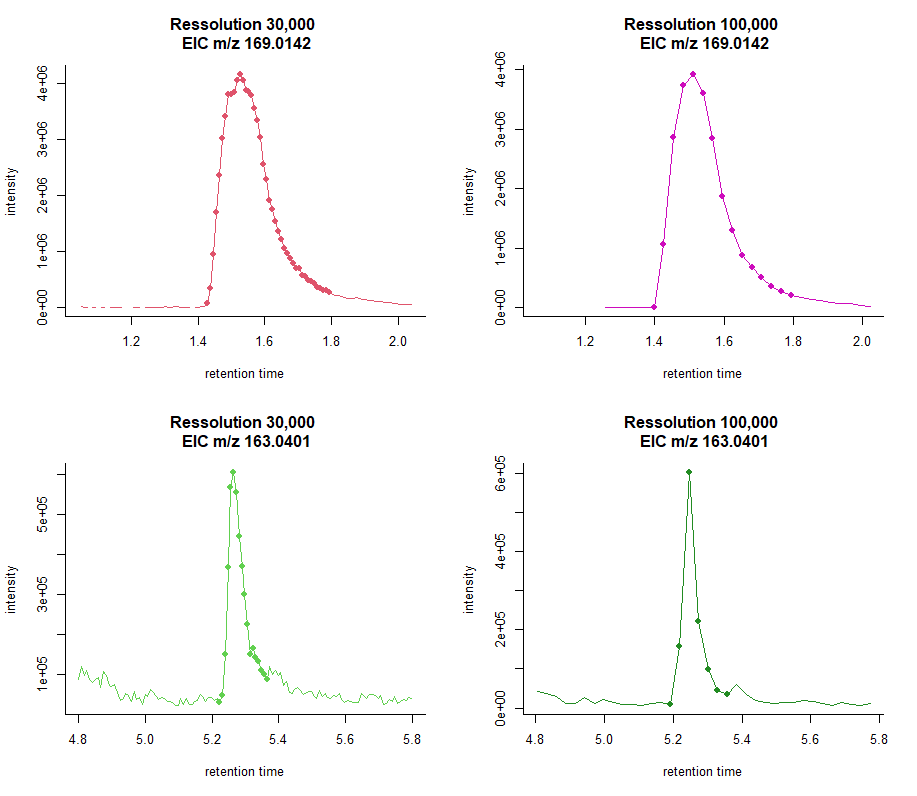
Mass resolution
Acquiring data at higher resolving power results in better mass accuracy, but the scan duration increases with increasing resolving power, thus diminish number of scan/peak
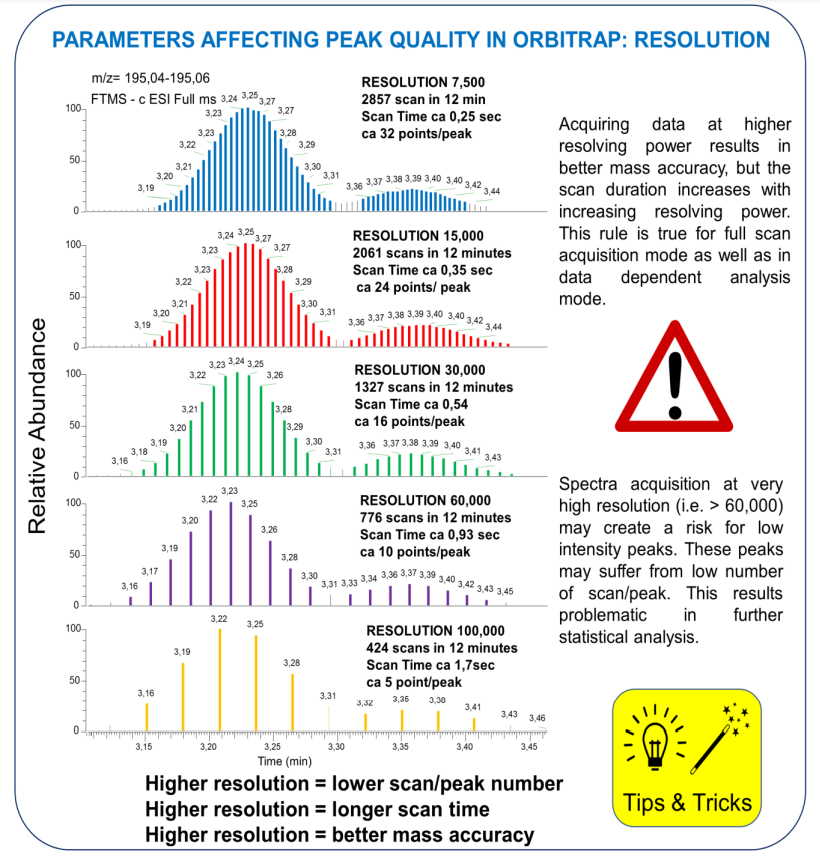
Mass resolution
Full Scan Mode
Only one MS function without induced fragmentation is acquired to generate ions of the molecular species, adducts and in-source fragments.
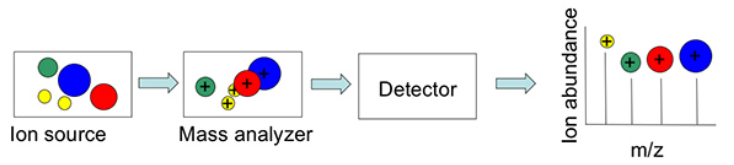
Full Scan Mode
Acquisition performed as a full scan over a wide m/z range, depending on the particular application and/or compounds of interest.
- Lipidomics: 100 - 2,000 Da
- Metabolomics on biofluids: 70-800 Da
Data Dependent Acquisition Mode
The DDA mode allows the production of fragmentation spectra for structure elucidation.
The instrument acquires a full scan MS1 and, when certain criteria are met, performs a specified number of MS/MS acquisitions on the most intense ions before switching back to full scan MS1.
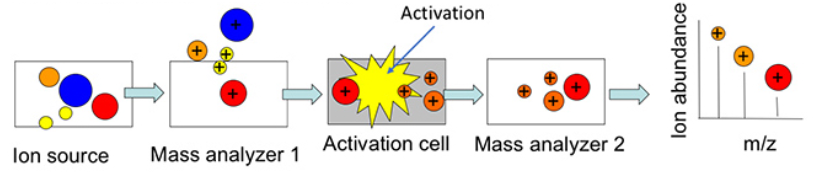
Data Dependent Acquisition Mode

Data Dependent Acquisition Mode

Data Dependent Acquisition Mode